Jmol
Jmol is computer software for molecular modelling chemical structures in 3-dimensions.[2] Jmol returns a 3D representation of a molecule that may be used as a teaching tool,[3] or for research e.g., in chemistry and biochemistry. It is written in the programming language Java, so it can run on the operating systems Windows, macOS, Linux, and Unix, if Java is installed. It is free and open-source software released under a GNU Lesser General Public License (LGPL) version 2.0. A standalone application and a software development kit (SDK) exist that can be integrated into other Java applications, such as Bioclipse and Taverna.
A popular feature is an applet that can be integrated into web pages to display molecules in a variety of ways. For example, molecules can be displayed as ball-and-stick models, space-filling models, ribbon diagrams, etc.[4] Jmol supports a wide range of chemical file formats, including Protein Data Bank (pdb), Crystallographic Information File (cif), MDL Molfile (mol), and Chemical Markup Language (CML).[5] There is also a JavaScript-only (HTML5) version, JSmol, that can be used on computers with no Java.[6]
The Jmol applet, among other abilities, offers an alternative to the Chime plug-in,[3] which is no longer under active development. While Jmol has many features that Chime lacks, it does not claim to reproduce all Chime functions, most notably, the Sculpt mode. Chime requires plug-in installation and Internet Explorer 6.0 or Firefox 2.0 on Microsoft Windows, or Netscape Communicator 4.8 on Mac OS 9. Jmol requires Java installation and operates on a wide variety of platforms. For example, Jmol is fully functional in Mozilla Firefox, Internet Explorer, Opera, Google Chrome, and Safari.
Jmol translations
Chen, Jim X. (2008), Springer, ed., Guide to Graphics Software Tools, p. 471, ISBN 978-1-84800-900-4
Herráez, A (2006), "Biomolecules in the Computer: Jmol to the Rescue", Biochemistry and Molecular Biology Education, 34 (4): 7, doi:10.1002/bmb.2006.494034042644
Herráez, A (2007), Lulu, ed., How to Use Jmol to Study and Present Molecular Structures, Volume 1, p. 21, ISBN 978-1-84799-259-8
Willighagen, E (2001), "Processing CML conventions in Java" (PDF), Internet Journal of Chemistry, 4 (4): 1–9[permanent dead link]
-------------------
Jmol returns a 3D representation of a molecule that may be
used as a
teaching tool, or for research e.g. in chemistry and biochemistry.
Features include:

---------------------
Installing:
mol three-dimensional structure rendering of streptavidin
|
|
| Developer(s) | Jmol development team |
|---|---|
| Initial release | 2001 |
| Stable release | 14.6.4 (October 15, 2016) [±] |
| Preview release | 14.5.0 (7 November 2015) [±] |
| Repository | sourceforge |
| Development status | Active |
| Written in | Java |
| Operating system | Cross-platform |
| Platform | Systems with Java and Web browsers without Java |
| Available in | 16 languages |
| Type | Molecular modelling |
| License | LGPL 2.0 |
| Website | www |
Jmol is computer software for molecular modelling chemical structures in 3-dimensions.[2] Jmol returns a 3D representation of a molecule that may be used as a teaching tool,[3] or for research e.g., in chemistry and biochemistry. It is written in the programming language Java, so it can run on the operating systems Windows, macOS, Linux, and Unix, if Java is installed. It is free and open-source software released under a GNU Lesser General Public License (LGPL) version 2.0. A standalone application and a software development kit (SDK) exist that can be integrated into other Java applications, such as Bioclipse and Taverna.
A popular feature is an applet that can be integrated into web pages to display molecules in a variety of ways. For example, molecules can be displayed as ball-and-stick models, space-filling models, ribbon diagrams, etc.[4] Jmol supports a wide range of chemical file formats, including Protein Data Bank (pdb), Crystallographic Information File (cif), MDL Molfile (mol), and Chemical Markup Language (CML).[5] There is also a JavaScript-only (HTML5) version, JSmol, that can be used on computers with no Java.[6]
The Jmol applet, among other abilities, offers an alternative to the Chime plug-in,[3] which is no longer under active development. While Jmol has many features that Chime lacks, it does not claim to reproduce all Chime functions, most notably, the Sculpt mode. Chime requires plug-in installation and Internet Explorer 6.0 or Firefox 2.0 on Microsoft Windows, or Netscape Communicator 4.8 on Mac OS 9. Jmol requires Java installation and operates on a wide variety of platforms. For example, Jmol is fully functional in Mozilla Firefox, Internet Explorer, Opera, Google Chrome, and Safari.
- Crystal structure of an H/ACA box RNP from Pyrococcus furiosus.
-
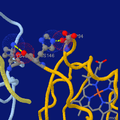 Highlighting two salt bridges in hemoglobin tetramer (hemo group as sticks at bottom-right).
Highlighting two salt bridges in hemoglobin tetramer (hemo group as sticks at bottom-right). -
 A fragment of transcription factor TFIIIA forming three consecutive zinc finger motifs, bound to a stretch of DNA.
A fragment of transcription factor TFIIIA forming three consecutive zinc finger motifs, bound to a stretch of DNA. -
 Eubacterial 70S Ribosome from Thermus thermophilus.
Eubacterial 70S Ribosome from Thermus thermophilus.



See also
- Chemistry Development Kit (CDK)
- Comparison of software for molecular mechanics modeling
- Jmol extension for MediaWiki
- List of molecular graphics systems
- Molecular graphics
- Molecule editor
- Proteopedia
- PyMOL
- SAMSON
References
- "JSmol". Retrieved 2015-11-02.
External links
| Wikimedia Commons has media related to Jmol. |
- Official website
- Willighagen, Egon; Howard, Miguel (June 2007). "Fast and Scriptable Molecular Graphics in Web Browsers without Java3D". doi:10.1038/npre.2007.50.1
Jmol
Jmol is an open-source Java viewer for three-dimensional chemical structures with features for chemicals, crystals, materials and biomolecules.| Jmol 14.2.13 |
|
Price Free to download Size 56.2MB License GNU LGPL Developer Jmol Development Team Website jmol.sourceforge.net
System Requirements
Support:Java Runtime Environment Handbook, FAQs, Wiki, Mailing Lists, SourceForge Project Page
Selected
Reviews:
|
- Applet, Application, and Systems Integration Component:
- The JmolApplet is a web browser applet that can be integrated into web pages. It is ideal for development of web-based courseware and web-accessible chemical databases. The JmolApplet provides an upgrade path for users of the Chime plug-in
- The Jmol application is a standalone Java application that runs on the desktop
- The JmolViewer can be integrated as a component into other Java applications
- High-performance 3D rendering with no hardware requirements
- Supports a wide range of molecular file formats:
- MOD - MDL / Elsevier / Symyx structure (classic version V2000)
- V3000 - MDL / Elsevier / Symyx structure (new version V3000)
- SDF - MDL / Elsevier / Symyx structure (multiple models)
- CTFile - MDL / Elsevier / Symyx chemical table (generic)
- CIF - Crystallographic Information File - standard from the International Union of Crystallography
- mmCIF - Macromolecular Crystallographic Information File - standard from the International Union of Crystallography
- CML - Chemical Markup Language
- PDB - Protein Data Bank - Research Collaboratory for Structural Bioinformatics
- XYZ - XYZ format, XMol file - Minnesota Supercomputer Institute
- XYZ+vib - XYZ format with added vibrational vector information
- XYZ-FAH - XYZ format for Folding@home
- MOL2 - Sybyl, Tripos
- Alchemy - Tripos
- CSF - Fujitsu CAChe chemical structure, now Fujitsu Sygress
- GAMESS - General Atomic and Molecular Electronic Structure System output (both US and UK variants) - Gordon Research Group, Iowa State University
- Gaussian - Gaussian 94/98/03 output - Gaussian, Inc.
- Cube - Gaussian, Inc.
- Ghemical - The Ghemical computational chemistry package
- MM1GP - Ghemical molecular mechanics file
- HIN - IN / HIV files from HyperChem - Hypercube, Inc.
- Jaguar - Schrodinger, LLC
- MOLPRO - Molpro output
- MOPAC - MOPAC 93/97/2002 output (public domain)
- MGF - MOPAC 2007 (v.7.101) graphf output (public domain)
- NWCHEM - NWChem output - Pacific Northwest National Laboratory
- odydata - Odyssey data - WaveFunction, Inc.
- xodydata - Odyssey XML data - WaveFunction, Inc.
- QOUT - Q-Chem, Inc.
- SHELX - Structural Chemistry Department, University of Göttingen (Germany)
- SMOL - Spartan data - Wavefunction, Inc.
- spinput - Spartan data - Wavefunction, Inc.
- GRO - Gromos87 format from GROMACS
- PQR - Modified pdb format including charge and radius
- Amber - The Amber package of molecular simulation programs
- JME - Java Molecular Editor - Peter Ertl
- CASTEP - The CASTEP software package, uses density functional theory
- FHI-aims - Full-potential / all-electron electronic structure theory with local orbitals - Fritz-Haber-Institut der Max-Planck-Gesellschaft
- VASP - VASP / VAMP / Vienna ab-initio simulation package
- DGrid - Miroslav Kohout, Max-Planck Institute
- ADF - ADF output - Amsterdam Density Functional
- XSD - Accelrys Materials Studio
- AGL - ArgusLab
- DFT - Wien2k
- AMPAC - AMPAC output - Semichem, Inc.
- WebMO - WebMO interface to computational chemistry packages
- Molden - Electron density / molecular orbitals
- PSI3 - Output files from the PSI3 suite of quantum chemical programs
- CRYSTAL - Output files from CRYSTAL, a computational tool for solid state chemistry and physics. Theoretical Chemistry Group, Univ. Torino, Italy.
- Animations
- Vibrations
- Surfaces
- Orbitals
- Support for unit cell and symmetry operations
- Schematic shapes for secondary structures in biomolecules
- Measurements
- Distance
- Angle
- Torsion angle
- Support for the RasMol/Chime scripting language
- JavaScript support library (Jmol.js)
- Exports to jpg, png, gif, ppm, pdf, POV-Ray, Gaussian, Maya, vrml, x3d, idtf, web page
- Fully internationalised - Multi-language:
- Translated into multiple languages: Catalan (ca), Chinese (both zh_CN and zh_TW) Czech (cs), Dutch (nl), French (fr), German (de), Hungarian (hu), Italian (it), Korean (ko), Portuguese - Brazil (pt_BR), Spanish (es), Turkish (tr), (in addition to the native American English, en-US, and British English, en-GB).
- Automatically adopts the language of the user's operating system, if it is among the translations available

---------------------
Installing:
No comments:
Post a Comment